Über Morbus Fabry
Morbus Fabry im Überblick
Was ist Morbus Fabry?
Morbus Fabry gehört zu den sogenannten lysosomalen Speicherkrankheiten, einer Form der Stoffwechselkrankheiten, und wird durch einen Enzymdefekt verursacht. Alternativ wird die Erkrankung auch mit Fabry-Krankheit, Fabry-Anderson-Krankheit oder Fabry-Syndrom bezeichnet.
Morbus Fabry ist erblich bedingt und verläuft bei Männern oft schwerer als bei Frauen. Insgesamt gibt es eine Vielzahl von Symptomen, die unterschiedlich stark ausgeprägt sein können und verschiedene Organe betreffen. Man spricht daher auch von einer Multisystem-Erkrankung. Unbehandelt schreitet die Erkrankung in der Regel immer weiter fort, doch mit der Enzymersatztherapie und der Chaperontherapie stehen den Betroffenen heute wirkungsvolle Behandlungsoptionen zur Verfügung. Wie diese Therapien funktionieren und welche therapeutischen Möglichkeiten außerdem bestehen, lesen Sie im Abschnitt Therapie.1
Wie viele Betroffene gibt es?
Morbus Fabry wird zu den Seltenen Erkrankungen gezählt, das heißt, es sind nicht mehr als fünf von 10.000 Menschen betroffen.2 Tatsächlich wurde zunächst geschätzt, dass nur eine von 40.000–170.000 Personen Morbus Fabry hat.3 In Deutschland haben etwa 1.000 Patient*innen die Diagnose Morbus Fabry erhalten, von denen ca. 800 Betroffene behandelt werden. Es wird jedoch davon ausgegangen, dass es deutlich mehr Menschen gibt, die mit Morbus Fabry leben.4,5 Die Erkrankung wird häufig nicht oder erst spät erkannt, weil die Symptome vielfältig sind und auch Merkmale anderer Krankheitsbilder sein können. Zudem gibt es nur wenige Mediziner*innen, die Erfahrung mit der Diagnostik des Morbus Fabry haben. Doch einige Kliniken haben sich auf die Diagnose und Therapie lysosomaler Speicherkrankheiten spezialisiert und verfügen über die notwendige Expertise; eine Übersicht hierzu finden Sie unter Fachkliniken.
Trotz der Seltenheit der Erkrankung gibt es Selbsthilfeorganisationen, die Ihnen die Möglichkeit geben, mit anderen Betroffenen in Kontakt zu kommen und von einem Erfahrungsaustausch zu profitieren.
Wodurch wird Morbus Fabry verursacht?
Bei Patient*innen mit Morbus Fabry ist ein bestimmter Abschnitt der Erbinformation, das sogenannte GLA-Gen, verändert. Diese Gen-Mutation führt dazu, dass ein Enzym mit dem Namen Alpha-Galaktosidase A (α-GalA) nicht oder nur teilweise funktionsfähig ist. α-GalA ist für den Abbau des Fettstoffs Globotriaosylceramid (Gb3) in den Lysosomen, dem Verdauungsorgan der Zellen, verantwortlich. Aufgrund der mangelnden bzw. fehlenden Aktivität von α-GalA kommt es zu einer fortschreitenden Ansammlung von Gb3 in den Endothelzellen verschiedener Organsysteme. Die genauen Zusammenhänge zwischen der Ablagerung von Gb3 in den Zellen und der Schädigung der betroffenen Organe sind bislang noch nicht endgültig geklärt. Einiges deutet darauf hin, dass verschiedene biochemische Mechanismen, u. a. Entzündungsprozesse, an den Organschädigungen und dem Verlauf der Erkrankung beteiligt sind.1,6
Wie wird Morbus Fabry vererbt?
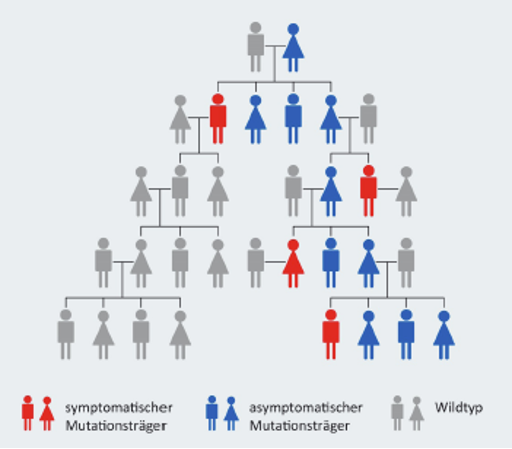
Erkrankungen, die auf einer Veränderung der Erbinformation beruhen, können vererbt werden. Somit ist Morbus Fabry ein Beispiel für eine Erbkrankheit. Die für die Erkrankung verantwortliche Mutation des GLA-Gens befindet sich auf dem X-Chromosom. Männer haben in ihren Zellen jeweils ein X- und ein Y-Chromosom. Ist das X-Chromosom von der Gen-Mutation betroffen, so erkranken diese Männer an Morbus Fabry. Frauen sind hingegen mit zwei X-Chromosomen ausgestattet, von denen eines nach dem Zufallsprinzip inaktiviert wird. Dass beide X-Chromosomen die Gen-Mutation tragen, ist extrem selten. Häufiger ist nur eines dieser beiden X-Chromosomen betroffen. Wie stark bei einer Frau die Erkrankung ausgeprägt ist, hängt davon ab, ob in den Zellen des jeweiligen Organs in der Mehrzahl intakte oder aber mutierte X-Chromosomen aktiv sind.7
Aufgrund dieses sogenannten X-chromosomalen Erbgangs spielt die Analyse des Familienstammbaums bei der Diagnose von Morbus Fabry sowie die genetische Beratung von Betroffenen und ihren Angehörigen eine wichtige Rolle. Wird bei einem*einer Patient*in Morbus Fabry diagnostiziert, können mithilfe eines Stammbaums weitere Verwandte identifiziert werden, die ebenfalls die Gen-Mutation tragen.
Wie groß die Wahrscheinlichkeit ist, dass die Erkrankung von einem betroffenen Elternteil an dessen Kinder weitergegeben wird, zeigt Abbildung 2.
Welche Formen von Morbus Fabry gibt es?
Klassischer Morbus Fabry
Bei der klassischen Form treten die ersten Symptome in der Regel bereits während der Kindheit auf. Mit zunehmendem Alter weiten sich die Beschwerden auf weitere Organe aus und im Erwachsenenalter kommt es vor allem zu Komplikationen am Herzen, der Niere oder dem Gehirn.3
Atypischer Morbus Fabry
Diese Form wird auch als Late-Onset-Form (englisch: spät beginnend) bezeichnet. Wie der Name andeutet, setzt die Erkrankung später ein als bei der klassischen Form, typischerweise im Alter zwischen 30 und 60 Jahren. Meist ist nur ein Organsystem, überwiegend das Herz, betroffen.7
Weitere Informationen zur klassischen und atypischen Form des Morbus Fabry sind unter Verlauf und Prognose für Sie zusammengestellt.
- Mehta A, Hughes DA. Fabry disease. In: Adam MP, Everman DB, Mirzaa GM, et al. (Hrsg.), GeneReviews(®) [Internet]. University of Washington, Seattle. Copyright © 1993-2022, Seattle (WA), 2002 [Updated 2022]
- Bundesgesundheitsministerium. Seltene Erkrankungen. 2022. https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html, abgerufen am: 28.09.2022
- Paim-Marques L, de Oliveira RJ, Appenzeller S. Multidisciplinary management of fabry disease: current perspectives. J Multidiscip Healthc 2022;15:485-95
- Information der führenden Experten (Key Opinion Leader). 2021. Advisory Board.
- IQVIA TMP Sonderstudie Morbus Fabry. 2020.
- Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30
- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 2018;123(4):416-27
Symptome
Wie äußert sich Morbus Fabry?
Morbus Fabry ist eine sogenannte Multisystem-Erkrankung, das heißt, dass viele unterschiedliche Gewebe und Organe betroffen sind. Demzufolge kann sich die Erkrankung mit einer Vielfalt an Symptomen präsentieren.
Die ersten Anzeichen treten bei der klassischen Form bereits während der Kindheit, im Alter zwischen drei und zehn Jahren, auf. Bei Mädchen setzen die Symptome durchschnittlich etwas später ein als bei Jungen.1 Mit zunehmendem Alter weiten sich gewöhnlich die Schäden an Geweben und Organen aus, sodass sich das Krankheitsbild im Laufe der Zeit verändert und es z. T. auch zu lebensbedrohlichen Komplikationen kommen kann.
Bei der atypischen Form treten keine oder nur wenige der im Folgenden beschriebenen charakteristischen Symptome des klassischen Morbus Fabry auf. Zumeist ist in diesen Fällen nur ein einzelnes Organsystem betroffen, besonders häufig ist es das Herz.1
Es ist wichtig zu wissen, dass sich die Erkrankung nicht immer auf die gleiche Weise präsentiert. Welche Krankheitszeichen in welcher Ausprägung auftreten, ist von Person zu Person unterschiedlich. Auch innerhalb einer Familie können die Symptome und der Verlauf verschieden sein.
Die ersten Anzeichen im Kindes- und Jugendalter
Schmerzen
Kribbeln und brennender Schmerz in den Händen und Füßen gehören zu den neurologischen Symptomen und stellen häufig die ersten Anzeichen der Erkrankung dar. Von dort aus können die Schmerzen in die Arme und Beine oder auch den restlichen Körper ausstrahlen. Die Schmerzen treten entweder dauerhaft, also chronisch, oder phasenweise auf. Diese Schmerz-Episoden, die auch Fabry-Krisen genannt werden, können durch körperliche Anstrengung, Fieber, Stress oder eine plötzliche Änderung der Umgebungstemperatur oder Luftfeuchtigkeit ausgelöst werden und dauern von wenigen Minuten bis zu mehreren Tagen an. Bei Kindern werden diese Symptome häufig falsch eingeschätzt und als Wachstumsschmerzen angesehen. Im Erwachsenenalter können die Schmerzen zurückgehen oder ganz verschwinden.1,2
Betroffene können den Schmerz-Episoden vorbeugen, indem körperliche Überanstrengung, Stress, Übermüdung und extreme Temperaturen vermieden werden. Bei großer Hitze können Klimaanlagen, Kühlwesten und kühlende Gesichtssprays hilfreich sein. Darüber hinaus ist es wichtig, ausreichend zu trinken.3,4
Haut
Kleine flache oder leicht erhobene rote oder lilafarbene Punkte auf der Haut sind ein typisches Merkmal bei Morbus Fabry. Etwa zwei von drei erkrankten Männern und eine von drei erkrankten Frauen sind von diesen Hautveränderungen betroffen5, die meist im Jugendalter erstmalig in Erscheinung treten. Diese in der Fachsprache Angiokeratome genannten Punkte bilden sich überwiegend im Bereich zwischen dem Bauchnabel und den Oberschenkeln, doch auch die Bindehaut des Auges oder der Innenbereich des Mundes können betroffen sein. Mit zunehmendem Alter werden die Angiokeratome in der Regel zahlreicher und größer.1,2
Neben der gut sichtbaren Hautveränderung der Angiokeratome stellen Patient*innen mit Morbus Fabry häufig fest, dass sie nicht oder nur eingeschränkt schwitzen können. Dieses Phänomen wird als Anhidrosis bzw. Hypohidrosis bezeichnet.1 Etwa die Hälfte der Männer und ein Viertel der Frauen mit Morbus Fabry sind von Hypohidrosis betroffen.5 In der Folge der dadurch gestörten Temperaturregulation sind diese Menschen besonders hitzeempfindlich.
Augen
Bei 70–80 von 100 Patient*innen lässt sich eine Cornea verticillata genannte Trübung der Hornhaut im Auge feststellen.6 Diese beeinträchtigt in der Regel nicht das Sehvermögen und kann nur mithilfe eines augenärztlichen Untersuchungsgerätes erkannt werden. Dabei zeigen sich weiße bis goldbraune Schlieren, die wirbelartig auf der Hornhaut angeordnet sind.2
Weitere Symptome der Augen sind eine Schlängelung der Blutgefäße in der Bindehaut und/oder Netzhaut sowie eine charakteristische Trübung der Linse, die auch als Fabry-Katarakt bezeichnet wird.1,2
Ohren
Die Ohren können ebenfalls bereits im Kindes- und Jugendalter betroffen sein.7 Mit zunehmendem Alter treten Hörprobleme dann immer öfter auf. So kann es zu einem fortschreitenden Gehörverlust oder Tinnitus kommen.7 Letzteres bezeichnet Ohrengeräusche wie ein Klingeln, Pfeifen, Summen oder Rauschen, das auf einem oder beiden Ohren auftritt.
Bei einer stärkeren Einschränkung der Hörfähigkeit können Hörgeräte die Lebensqualität der Betroffenen verbessern.3,4
Verdauungstrakt
Morbus Fabry kann sich auch auf den Magen und den Darm auswirken und Symptome wie Durchfall, Übelkeit und Erbrechen hervorrufen. Daher haben Betroffene manchmal Schwierigkeiten, an Gewicht zuzunehmen. Auch Schmerzen im Unterleib, die insbesondere nach dem Essen auftreten, können mit der Erkrankung in Zusammenhang stehen.1 Diese Symptome beginnen ebenfalls typischerweise während der Kindheit und bleiben im weiteren Verlauf der Erkrankung bestehen.8
Eine Umstellung der Ernährung auf mehrere kleinere Mahlzeiten, die reich an Ballaststoffen sind, kann Betroffenen helfen, die Symptome zu lindern.4
Spätere Symptome und Komplikationen
Niere
Eine Beeinträchtigung der Niere beginnt häufig im Alter zwischen 10 und 30 Jahren und äußert sich durch eine erhöhte Menge an Eiweiß im Urin (Proteinurie). Im Krankheitsverlauf kommt es zunehmend zu Gewebeschäden, die in einer schrittweisen Abnahme der Nierenfunktion resultieren. Dies kann bis hin zu einem vollständigen Versagen der Nierenfunktion führen, was eine regelmäßige Dialyse oder Nierentransplantation erforderlich macht. Bei Männern tritt dies durchschnittlich im Alter von 30 bis 60 Jahren ein und ist eine der häufigsten Komplikationen bei Morbus Fabry.1
Die Funktion der Nieren sollte bei Patient*innen mit Morbus Fabry regelmäßig überprüft werden. Liegt eine Beeinträchtigung vor, so können Betroffene durch eine salz- und eiweißarme Ernährung die Behandlung unterstützen.3
Herz
Komplikationen, die das Herz betreffen, zählen zu den häufigsten Beschwerden bei Erwachsenen mit Morbus Fabry, insbesondere bei der atypischen Form.8 Beispielsweise kann sich die Struktur des Herzmuskels verändern, was sich negativ auf die Pumpleistung des Herzens auswirkt. Außerdem können Herzrhythmusstörungen, d. h. ein zu schneller, zu langsamer oder unregelmäßiger Herzschlag, auftreten. Auch anfallsartige Schmerzen bzw. ein Druckgefühl im Brustkorb (Angina pectoris) gehören zu den möglichen Komplikationen.1,9 Eine Beeinträchtigung des Herzens äußert sich anfangs häufig durch eine Einschränkung der körperlichen Leistungsfähigkeit, Müdigkeit und Luftnot bei Anstrengung.
Gehirn
Kopfschmerzen und Schwindel gehören zu den leichteren Symptomen, die das Gehirn betreffen. Schwerwiegender ist die sogenannte transitorische ischämische Attacke (TIA) und der Schlaganfall. Letzterer ist lebensbedrohlich und tritt bei Patient*innen mit Morbus Fabry deutlich häufiger auf als in der Allgemeinbevölkerung.1 TIA ist der medizinische Fachausdruck für eine plötzlich auftretende, vorübergehende Durchblutungsstörung des Gehirns. Im Gegensatz zum Schlaganfall, der ebenfalls durch eine gestörte Blutversorgung verursacht wird, gehen bei der TIA die Symptome innerhalb von 24 Stunden zurück und es bleiben keine dauerhaften Schäden bestehen.9
Die Symptome wirken sich auf die Lebensqualität aus
Die oben beschriebenen Symptome können sich allein oder in Kombination erheblich auf das Leben und den Alltag der Betroffenen auswirken. So kann die körperliche und schulische Leistungsfähigkeit von Kindern und Jugendlichen sowie deren Teilhabe am gesellschaftlichen Leben beeinträchtigt sein.1 Auch bei erwachsenen Männern und Frauen beeinflusst Morbus Fabry die Lebensqualität, wobei sich Schmerzen und Komplikationen besonders stark auswirken.10,11
Unterscheiden sich die Symptome von Männern und Frauen?
Grundsätzlich gilt, dass sich Morbus Fabry immer sehr individuell äußert. Aufgrund des Vererbungsmusters sind die Symptome in ihrer Art und Ausprägung bei Frauen jedoch besonders vielfältig. So können Frauen, die die Krankheit geerbt haben, vollständig ohne Krankheitszeichen bleiben, ebenso stark wie Männer mit der klassischen Form betroffen sein oder aber jede Zwischenstufe davon einnehmen. Typischerweise erscheinen die ersten Anzeichen bei Mädchen später als bei Jungen und auch Komplikationen der lebenswichtigen Organe treten bei Frauen durchschnittlich zehn Jahre später auf als bei Männern.1
- Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30
- Mehta A, Hughes DA. Fabry disease. In: Adam MP, Everman DB, Mirzaa GM, et al. (Hrsg.), GeneReviews(®) [Internet]. University of Washington, Seattle. Copyright © 1993-2022, Seattle (WA), 2002 [Updated 2022]
- Morbus Fabry Selbsthilfegruppe e.V. Morbus Fabry Broschüre. https://fabry-shg.org/informationsmaterial-eigenes/, abgerufen am: 28.09.2022
- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 2018;123(4):416-27
- Orteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry outcome survey. Br J Dermatol 2007;157(2):331-7
- Sodi A, Ioannidis AS, Mehta A, et al. Ocular manifestations of Fabry's disease: data from the Fabry Outcome Survey. Br J Ophthalmol 2007;91(2):210-4
- Keilmann A, Hajioff D, Ramaswami U. Ear symptoms in children with Fabry disease: data from the Fabry Outcome Survey. J Inherit Metab Dis 2009;32(6):739
- Paim-Marques L, de Oliveira RJ, Appenzeller S. Multidisciplinary management of fabry disease: current perspectives. J Multidiscip Healthc 2022;15:485-95
- IQWIG. 2022. https://www.gesundheitsinformation.de, abgerufen am: 05.10.2022
- Arends M, Körver S, Hughes DA, et al. Phenotype, disease severity and pain are major determinants of quality of life in Fabry disease: results from a large multicenter cohort study. J Inherit Metab Dis 2018;41(1):141-9
- Andonian C, Beckmann J, Mayer O, et al. Quality of life in patients with Fabry's disease: a cross-sectional study of 86 adults. Cardiovasc Diagn Ther 2022;12(4):426-35
Diagnose
Wie wird Morbus Fabry festgestellt?
Bei Morbus Fabry handelt es sich um eine seltene Erkrankung, die in der Regel in spezialisierten Fachzentren diagnostiziert und behandelt wird. Zudem kann das Krankheitsbild von Person zu Person stark variieren und umfasst eine ganze Reihe an möglichen Symptomen, die ebenfalls bei anderen – häufigeren – Erkrankungen auftreten. Aus diesen Gründen kann es ab Beginn der Symptome oft einige Jahre (durchschnittlich vier Jahre bei Kindern und 10,5 Jahre bei Erwachsenen) dauern, bis Morbus Fabry diagnostiziert wird.6 Darüber hinaus wird bei einem von vier Fällen zunächst eine falsche Diagnose gestellt.7 Dies kann für die Betroffenen eine schwierige Zeit mit belastenden körperlichen und seelischen Beeinträchtigungen sein.
Morbus Fabry kann sich durch eine Vielzahl unterschiedlicher Krankheitszeichen äußern, die ausführlich unter der Rubrik Symptome beschrieben werden. Doch einige Beschwerden sind besonders häufig und sollten den*die Ärzt*in an diese Stoffwechselkrankheit denken lassen.
Durch Aktivieren des Videos willigen Sie in die Übermittlung Ihrer Daten an Google Ireland Limited sowie gegebenenfalls an ein Drittland ein.
Mehr erfahren
Video laden Youtube immer entsperren
Krankheitszeichen bei Kindern und Jugendlichen1,2
- Kleine rote oder lilafarbene, punktförmige Hautveränderungen, meist im Bereich zwischen dem Bauchnabel und den Oberschenkeln
- Brennende Schmerzen in den Händen und Füßen
- Eingeschränkte oder fehlende Fähigkeit zu Schwitzen
- Beschwerden des Verdauungstraktes wie Durchfall und Bauchschmerzen
- Empfindlichkeit gegenüber Kälte und Hitze
- Starke Müdigkeit und Abgeschlagenheit (Fatigue)
Krankheitszeichen bei Erwachsenen2
- Herzprobleme ohne erkennbare Ursache, z. B. eine Vergrößerung des Herzmuskels der linken Herzkammer oder Herzrhythmusstörungen
- Schlaganfall oder transitorische ischämische Attacke ohne erkennbare Ursache
- Funktionseinschränkung der Niere ohne erkennbare Ursache
Wie wird der Verdacht auf Morbus Fabry bestätigt?
Wurde die Fabry-Krankheit einmal in Betracht gezogen, so ist der endgültige Nachweis recht einfach. Die Diagnostik umfasst zum einen den Enzymaktivitätstest, bei dem die Aktivität des Enzyms Alpha-Galaktosidase A (α-GalA) bestimmt wird. Ist keine oder nur eine minimale Aktivität (< 1 %) feststellbar, handelt es sich um Morbus Fabry der klassischen Form. Bei einer Restaktivität von 1–30 % liegt ein atypischer Morbus Fabry vor.3 Zum anderen wird mithilfe eines Gentests untersucht, ob eine Veränderung des GLA-Gens vorliegt. Welches Nachweisverfahren erforderlich ist, unterscheidet sich bei Männern und Frauen. Die Diagnostik wird heute immer häufiger durch eine Messung der Lyso-Gb3-Menge ergänzt. Lyso-Gb3 ist eine Form des Fettstoffs, der sich aufgrund des Enzymdefekts bei Menschen mit Morbus Fabry ansammelt. Die Bestimmung seiner Konzentration im Blut kann die Erkennung der Erkrankung und nachfolgend die Verlaufsbeobachtung unterstützen.2
Diagnose bei Männern
Bei Männern ist der α-GalA-Aktivitätstest theoretisch ausreichend, um eine sichere Diagnose stellen zu können. Trotzdem wird empfohlen, zusätzlich einen Gentest durchzuführen, um die Art der Mutation des GLA-Gens herauszufinden.4 Zudem kann in Einzelfällen auch bei Männern die Enzymaktivität in der Diagnostik im Normalbereich liegen, obwohl ein Morbus Fabry vorliegt. Auch aus diesem Grund kann ein Gentest sinnvoll sein.
Diagnose bei Frauen
Da bei Frauen die Aktivität von α-GalA im Normalbereich liegen kann, reicht ein Aktivitätstest nicht aus. Vielmehr ist immer eine Analyse des Erbguts notwendig.4
Welche Eingriffe sind für die Diagnosestellung erforderlich?
Sowohl der Aktivitätstest als auch der Gentest und die Messung der Lyso-Gb3-Menge können anhand einer einzigen Blutprobe durchgeführt werden. Besonders schnell und einfach sind sogenannte Trockenbluttests. Hierfür werden wenige Bluttropfen aus der Fingerkuppe auf ein spezielles Filterpapier aufgebracht. Nach der Trocknung wird die Probe an ein Labor verschickt und dort untersucht.5 Solche Trockenbluttests sind für die Patient*innen kostenfrei.
Was passiert nach der Diagnose?
Nachdem die Diagnose Fabry-Krankheit mit den oben beschriebenen Verfahren gestellt wurde, sollte jede betroffene Person umfassend untersucht werden. Dabei sollten alle typischerweise beteiligten Organe berücksichtigt werden, um ein vollständiges Bild des aktuellen Gesundheitszustands zu erhalten. Auf dieser Grundlage wird vom Behandlungsteam gemeinsam mit dem*der Patient*in das weitere therapeutische Vorgehen geplant. Nähere Informationen zu den Behandlungsoptionen sind unter Therapie zusammengefasst.2
Da es sich bei Morbus Fabry um eine Erbkrankheit handelt, sollte bei neu diagnostizierten Fällen auch immer eine Stammbaumanalyse durchgeführt und das Erbgut der anderen Familienmitglieder untersucht werden. So können weitere Betroffene identifiziert werden und von einer Behandlung profitieren.8
- Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clinical Genetics 2019;96(2):107-17
- Mehta A, Hughes DA. Fabry disease. In: Adam MP, Everman DB, Mirzaa GM, et al. (Hrsg.), GeneReviews(®) [Internet]. University of Washington, Seattle. Copyright © 1993-2022, Seattle (WA), 2002 [Updated 2022]
- Paim-Marques L, de Oliveira RJ, Appenzeller S. Multidisciplinary management of fabry disease: current perspectives. J Multidiscip Healthc 2022;15:485-95
- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 2018;123(4):416-27
- Arndt T. Trockenblut. In: Gressner AM, Arndt T (Hrsg.), Lexikon der Medizinischen Laboratoriumsdiagnostik. Springer Berlin Heidelberg, Berlin, Heidelberg, 2019;2363
- Reisin R, Perrin A, García-Pavía P. Time delays in the diagnosis and treatment of Fabry disease. Int J Clin Pract 2017;71(1)
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004;34(3):236-42
- Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30
Verlauf & Prognose
Wie schreitet die Erkrankung voran?
Bei Morbus Fabry handelt es sich um eine sogenannte progrediente, d. h. fortschreitende, Erkrankung. Durch die fehlende oder unzureichende Aktivität des Enzyms Alpha-Galaktosidase A (α-GalA) kann der Fettstoff Globotriaosylceramid (Gb3) nicht abgebaut werden und sammelt sich in den Zellen an. Unbehandelt setzt sich dies immer weiter fort. So werden zunächst die Abläufe in Zellen und schließlich in ganzen Geweben beeinträchtigt. Im Laufe der Zeit reichern sich irreparable Schäden an, welche die Funktion von Organen behindern und in schweren Fällen zum Versagen einzelner oder mehrerer Organe führen können.1,2 Aufgrund dessen ist die durchschnittliche Lebenserwartung von Menschen mit Morbus Fabry gegenüber der Allgemeinbevölkerung reduziert.3,4 Es bestehen jedoch Behandlungsmöglichkeiten, mit deren Hilfe der Krankheitsverlauf gebremst, die Häufigkeit von Komplikationen vermindert und damit die Prognose sowie die Lebenserwartung verbessert werden können.5,6
Welche Rolle spielt eine Beeinträchtigung der Organe?
Die zunehmenden Schäden in Zellen und Geweben können die Funktion der betroffenen Organe beeinträchtigen und Komplikationen hervorrufen. Am häufigsten sind das Herz (z. B. Veränderungen der Herzmuskelstruktur, Herzrhythmusstörungen), die Nieren (chronische Nierenerkrankung) und das Gehirn (z. B. transitorische ischämische Attacken, Schlaganfall) betroffen.2 Ausführlich werden die Beeinträchtigungen dieser Organe unter Symptome beschrieben.
Daneben leiden Menschen mit Morbus Fabry auch häufig unter einer Depression. Studien zufolge sind bis zu sechs von zehn Personen davon betroffen.7 Ein oft langer Leidensweg bis zur Diagnose, körperliche Beschwerden, darunter vor allem Schmerzen, und Zukunftsängste tragen wesentlich zur Entstehung einer Depression bei. Erste Anzeichen dieser psychischen Erkrankung können unter anderem anhaltende Antriebsstörungen, erhöhte Reizbarkeit und ein Gefühl der Hoffnungslosigkeit sein.8
Die Beeinträchtigungen der Organe müssen im Rahmen einer ganzheitlichen Therapie gezielt behandelt und bei den Kontrolluntersuchungen regelmäßig begutachtet werden.
Was unterscheidet den klassischen und atypischen Verlauf?
Bei Morbus Fabry werden zwei Verlaufsformen unterschieden: die klassische und die atypische Form.
| Klassischer Morbus Fabry | Atypischer Morbus Fabry |
|---|---|
|
|
Der Verlauf des Morbus Fabry unterscheidet sich darüber hinaus auch zwischen den Geschlechtern. Welche Besonderheiten der Krankheitsverlauf von Frauen aufweisen kann, wird unter Symptome beschrieben.
Wie wird der Krankheitsverlauf beobachtet?
Um ein Fortschreiten der Erkrankung frühzeitig festzustellen und die Behandlung rechtzeitig anpassen zu können, sind für Menschen mit Morbus Fabry regelmäßige Kontrolluntersuchungen wichtig. Dabei werden unter anderem Hautveränderungen, Schmerzen, Beschwerden des Verdauungstrakts und psychologische Symptome erfasst. Daneben wird ein besonderes Augenmerk auf die Untersuchung von Herz, Nieren und Hirngefäßen gelegt. Häufig eingesetzte Untersuchungsmethoden sind die Elektrokardiografie (EKG), die Echokardiografie (ECHO), Blut- und Urin-Analysen sowie Ultraschall und Magnetresonanztomografie (MRT), die in der Regel ambulant und weitestgehend schmerzfrei durchgeführt werden können. Bei betroffenen Jungen und Männern wird empfohlen, die Untersuchungen einmal jährlich zu wiederholen, bei Mädchen und Frauen können die Abstände einzelner Untersuchungen auf zwei bis drei Jahre verlängert werden. Erhalten die Patient*innen eine Enzymersatztherapie, so wird eine häufigere – halbjährliche – Verlaufskontrolle empfohlen.12,13
Gibt es Besonderheiten in bestimmten Lebensphasen?
Morbus Fabry bei Kindern
Der Erkrankungsprozess, d. h. die Ansammlung des Fettstoffs in den Zellen, setzt bereits während der Entwicklung des Fötus ein. Die ersten Symptome treten bei der klassischen Form des Morbus Fabry im Kindesalter auf. Zu den typischen Krankheitszeichen bei Kindern gehören rote oder lilafarbene punktförmige Hautveränderungen, brennende Schmerzen in Händen und Füßen, eine eingeschränkte oder fehlende Fähigkeit zu Schwitzen, eine Empfindlichkeit gegenüber Hitze sowie Beschwerden des Verdauungstrakts. Da diese Symptome auch bei anderen Krankheiten auftreten können, sind Fehldiagnosen häufig und die Zeitspannen bis zur Diagnose des Morbus Fabry lang. So dauert es bei Kindern durchschnittlich vier Jahre vom Beginn der Symptome bis zur korrekten Diagnosestellung.14 Dabei ist ein frühzeitiger Therapiebeginn wichtig, um das Fortschreiten der Erkrankung zu verlangsamen bzw. zu verhindern und spätere irreparable Schäden und lebensbedrohliche Komplikationen zu vermeiden.10
Es ist wichtig zu wissen, dass wie bei Erwachsenen körperliche Belastung, Hitze oder starke Temperaturschwankungen einige der Beschwerden verstärken können.8 Darüber hinaus kann die Krankheitsdiagnose selbst sowie die eingeschränkte Teilhabe an sozialen und sportlichen Aktivitäten eine große psychische Last für die Kinder darstellen und die Lebensqualität einschränken.13 Viele Gespräche sowie die Aufklärung der Kinder und ihres Umfelds über die Erkrankung helfen, diese Last abzubauen.8
Schwangerschaft mit Morbus Fabry
Untersuchungen haben gezeigt, dass Morbus Fabry die Fruchtbarkeit von Männern und Frauen nicht beeinträchtigt.15 Somit ist die Erfüllung eines Kinderwunsches auch für Paare möglich, bei denen eine Morbus-Fabry-Erkrankung vorliegt. Im Rahmen einer genetischen Beratung können sich betroffene Paare darüber informieren, wie groß die Wahrscheinlichkeit ist, dass ihre Kinder die Erbanlage für Morbus Fabry erhalten.
Darüber hinaus ist es gut zu wissen, dass sich bei betroffenen Frauen einige Symptome der Erkrankung während der Schwangerschaft verstärken können. Dazu gehören Beschwerden des Verdauungstrakts, Schmerzen in den Händen und Füßen, Eiweiß im Urin und Kopfschmerzen. Außerdem kommt ein schwangerschaftsbezogener Bluthochdruck bei Schwangeren mit Morbus Fabry häufiger vor als in der Allgemeinbevölkerung.16 Das Behandlungsteam sollte in jedem Fall vor einer geplanten Schwangerschaft über den Kinderwunsch informiert werden, damit Medikamente, die zur Behandlung des Morbus Fabry zum Einsatz kommen und Missbildungen des Fötus hervorrufen könnten, rechtzeitig abgesetzt werden. Beispielsweise wird die Anwendung der Chaperontherapie während einer Schwangerschaft nicht empfohlen, da keine ausreichenden Daten hierzu vorliegen.17 Auch über die Fortsetzung oder Unterbrechung der Enzymersatztherapie sollte mit dem*der Ärzt*in beraten werden.11
- Eng CM, Fletcher J, Wilcox WR, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis 2007;30(2):184-92
- Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30
- Vedder AC, Linthorst GE, van Breemen MJ, et al. The Dutch Fabry cohort: diversity of clinical manifestations and Gb3 levels. J Inherit Metab Dis 2007;30(1):68-78
- Waldek S, Patel MR, Banikazemi M, et al. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet Med 2009;11(11):790-6
- Beck M, Hughes D, Kampmann C, et al. Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: a Fabry Outcome Survey analysis. Mol Genet Metab Rep 2015;3:21-7
- Germain DP, Charrow J, Desnick RJ, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet 2015;52(5):353-8
- Bolsover FE, Murphy E, Cipolotti L, et al. Cognitive dysfunction and depression in Fabry disease: a systematic review. J Inherit Metab Dis 2014;37(2):177-87
- Morbus Fabry Selbsthilfegruppe e.V. Morbus Fabry Broschüre. https://fabry-shg.org/informationsmaterial-eigenes/, abgerufen am: 28.09.2022
- Paim-Marques L, de Oliveira RJ, Appenzeller S. Multidisciplinary management of fabry disease: current perspectives. J Multidiscip Healthc 2022;15:485-95
- Ellaway C. Paediatric Fabry disease. Transl Pediatr 2016;5(1):37-42
- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 2018;123(4):416-27
- Mehta A, Hughes DA. Fabry disease. In: Adam MP, Everman DB, Mirzaa GM, et al. (Hrsg.), GeneReviews(®) [Internet]. University of Washington, Seattle. Copyright © 1993-2022, Seattle (WA), 2002 [Updated 2022]
- Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet 2019;96(2):107-17
- Reisin R, Perrin A, García-Pavía P. Time delays in the diagnosis and treatment of Fabry disease. Int J Clin Pract 2017;71(1)
- Hauser AC, Gessl A, Harm F, et al. Hormonal profile and fertility in patients with Anderson-Fabry disease. Int J Clin Pract 2005;59(9):1025-8
- Holmes A, Laney D. A retrospective survey studying the impact of Fabry disease on pregnancy. JIMD Rep 2015;21:57-63
- Fachinfo-Service. 2022. www.fachinfo.de, abgerufen am: 28.10.2022
Therapie
Welche Behandlungsmöglichkeiten gibt es für Morbus Fabry?
Morbus Fabry ist bis heute nicht heilbar, doch es gibt Therapien, die Beschwerden verringern und ein Fortschreiten der Krankheit verlangsamen bzw. verhindern können. Je früher die Diagnose erfolgt und damit auch die Therapie beginnt, umso besser sind die Chancen diese Ziele zu erreichen. Zudem sind regelmäßige Kontrollen erforderlich, um die Wirkung der Behandlung beurteilen und, falls nötig, Anpassungen vornehmen zu können.1 Welche Untersuchungen dazu durchgeführt werden und wie oft die Kontrolltermine erfolgen sollten, können Sie unter Verlauf und Prognose nachlesen.
Bei der Therapie werden in der Regel zwei Herangehensweisen miteinander kombiniert: zum einen die Behandlung der Krankheitsursache (Enzymersatztherapie, Chaperontherapie) und zum anderen die Behandlung der Beschwerden und möglicher Komplikationen (Begleittherapie).
Was sind die Ziele der Behandlung?
Im Allgemeinen werden mit der Therapie mehrere Ziele verfolgt. So sollen Symptome gelindert und bleibende Organschäden verhindert werden. Damit wird eine Verbesserung der Lebensqualität und nicht zuletzt eine Verlängerung der Lebenserwartung angestrebt. Je nach der Verlaufsform und dem Stadium der Erkrankung sowie der individuellen Situation der Patient*innen sollten die Ziele jedoch ganz persönlich angepasst werden.1
Da die Symptome, Folgeerkrankungen und Komplikationen von Morbus Fabry viele Organe betreffen können, besteht das Behandlungsteam in den Fabry-Zentren aus Expert*innen unterschiedlicher Fachrichtungen. Diese haben sich beispielsweise auf Erkrankungen des Herzens, der Nieren, der Nerven oder des Verdauungstraktes spezialisiert. Auch Fachkräfte aus dem Gebiet der Psychologie und Genetik gehören zu einem solchen multidisziplinären Team.2 Gemeinsam mit dem*der Patient*in und ggf. Angehörigen wird über die Therapiemöglichkeiten beraten und ein Behandlungsplan erstellt. Dabei sollten sowohl die Vorteile als auch mögliche Herausforderungen einer Therapie berücksichtigt werden.1
Was ist eine Enzymersatztherapie?
Die Enzymersatztherapie (EET) ist eine tragende Säule bei der Behandlung von Menschen mit Morbus Fabry. Sie bekämpft direkt die Ursache der Erkrankung, d. h. den Mangel am Enzym Alpha-Galaktosidase A (α-GalA). Patient*innen mit Morbus Fabry weisen eine unzureichende oder sogar fehlende Aktivität dieses Eiweißes auf, das für den Abbau bestimmter Fettstoffe (Gb3 und Lyso-Gb3) in den Zellen verantwortlich ist. Nähere Informationen hierzu können unter Fabry im Überblick nachgelesen werden. Bei der EET erhalten die Betroffenen eine künstlich hergestellte, funktionstüchtige α-GalA. Diese sorgt dafür, dass eine weitere Ansammlung der Fettstoffe verhindert und bereits abgelagertes Gb3 und Lyso-Gb3 reduziert wird.3
Für Patient*innen mit Morbus Fabry sind in Europa seit 2001 drei ERTs zugelassen. Studien zeigten, dass eine EET die Beschwerden reduzieren, die Lebensqualität verbessern, ein Fortschreiten der Krankheit verlangsamen und das Risiko für Komplikationen senken kann.4-10 Auch betroffene Kinder können von einer EET profitieren.11,12
Welche Besonderheiten gibt es bei der Anwendung der EET?
Die EET muss ein Leben lang angewendet werden, da bei einer Unterbrechung das funktionstüchtige Enzym fehlen und sich die Fettstoffe erneut ansammeln würden. Das künstlich hergestellte Enzym wird per Infusion in eine Vene gegeben. Die Infusion dauert zwischen 40 Minuten und vier Stunden und muss alle zwei Wochen wiederholt werden.13 Es ist wichtig, diesen Rhythmus einzuhalten und keine Infusion auszulassen, da der Wirkstoff im Körper abgebaut wird und das Enzym dem*der Patient*in bei einer Therapieunterbrechung fehlen würde. Derzeit wird untersucht, ob eine neue EET in einer höheren Dosierung im Abstand von vier Wochen verabreicht werden kann.14 Die ersten Infusionen werden in der Regel in spezialisierten Behandlungszentren verabreicht, anschließend kann die Behandlung in der örtlichen Klinik oder der Hausarztpraxis durchgeführt werden. Sogar eine Infusion zu Hause ist möglich. Bei dieser sogenannten Heiminfusion wird das Arzneimittel nach Hause geliefert und dort durch eine speziell ausgebildete medizinische Fachkraft verabreicht. In Einzelfällen ist es möglich, dass Patient*innen oder Angehörige nach einer ausführlichen Einweisung die Infusion selbstständig durchführen.13 Die Möglichkeit der Heiminfusion bietet den Betroffenen zahlreiche Vorteile und trägt zur Verbesserung der Lebensqualität bei.15
Welche Nebenwirkungen können bei der EET auftreten?
Medizinische Behandlungen sind immer auch mit einem Risiko für unerwünschte Reaktionen oder Nebenwirkungen verbunden. Bei der EET werden am häufigsten (bei mehr als einer von zehn Personen) sogenannte infusionsbedingte Reaktionen beobachtet.16,17 Diese können während der Infusion oder kurz danach auftreten. Häufig werden sie innerhalb der ersten zwei bis vier Monate nach Beginn der EET beobachtet. Zu diesen Reaktionen gehören Fieber, Schüttelfrost, Kopfschmerzen, Übelkeit, Erbrechen, Husten, Atembeschwerden und Müdigkeit. In den meisten Fällen sind diese Nebenwirkungen leicht bis mäßig ausgeprägt. In manchen Fällen kann es aber auch zu schwereren unerwünschten Reaktionen kommen. Infusionsbedingten Reaktionen kann mithilfe von Medikamenten, die vor oder während der Infusion gegeben werden, sowie einer Reduktion der Infusionsgeschwindigkeit vorgebeugt werden. Neben den infusionsbedingten Reaktionen können bei der EET weitere Nebenwirkungen auftreten, die meist gut behandelbar sind. Betroffene sollten sich diesbezüglich an ihr Behandlungsteam wenden.
Eine Herausforderung der EET stellen sogenannte neutralisierende Antikörper dar. Hierbei handelt es sich um Abwehrstoffe des Immunsystems, die in manchen Fällen als Reaktion auf die künstlich hergestellte α-GalA vom Körper produziert werden. Die neutralisierenden Antikörper verursachen zwar keine neuen Beschwerden, sie können aber die Wirkung der EET herabsetzen. Bei Männern mit der klassischen Form des Morbus Fabry, die keine körpereigene α-GalA produzieren, ist das Risiko, dass sich neutralisierende Antikörper bilden, am größten.18
Was ist eine Chaperontherapie?
Die Chaperontherapie bietet für bestimmte Patient*innen eine alternative Möglichkeit, die Krankheitsursache direkt zu behandeln. Bei manchen Menschen mit Morbus Fabry ist das Erbgut, also der Bauplan für das Enzym α-GalA, so verändert, dass sich das Eiweiß nicht in seine korrekte dreidimensionale Struktur falten kann. Dadurch wird dessen Transport vom Herstellungsort in der Zelle zu den Lysosomen verhindert und es kann dort nicht seine Aufgabe erfüllen. Bei der Chaperontherapie bindet der Wirkstoff, das Chaperon, an die α-GalA, korrigiert deren Fehlfaltung oder stabilisiert sie. So kann das Enzym in die Lysosomen transportiert werden, wo sich das Chaperon ablöst und die α-GalA ihre Funktion ausführen kann.19
Die Chaperontherapie ist in Europa seit 2016 für Patient*innen mit einem Mindestalter von zwölf Jahren zugelassen. Sie kann jedoch nur bei Betroffenen mit bestimmten Mutationen im Gen der α-GalA eingesetzt werden.20 Ob eine solche Mutation vorliegt, wird mithilfe einer Genanalyse untersucht, welche im Rahmen der Diagnosestellung durchgeführt wird.
Welche Besonderheiten gibt es bei der Anwendung der Chaperontherapie?
Ebenso wie die EET muss die Chaperontherapie ein Leben lang angewendet werden, da bei einer Beendigung das funktionstüchtige Enzym fehlen und sich die Fettstoffe erneut ansammeln würden. Anders als bei der EET handelt es sich bei der Chaperontherapie aber um eine Behandlung in Form von Kapseln. Dabei wird jeden zweiten Tag zur gleichen Uhrzeit eine Kapsel eingenommen. Zusätzlich ist zu beachten, dass die Einnahme nicht innerhalb von zwei Stunden vor oder nach einer Mahlzeit erfolgen soll.16
Welche Nebenwirkungen können bei der Chaperontherapie auftreten?
Die häufigsten Nebenwirkungen der Chaperontherapie sind Kopfschmerzen. Etwa jede zehnte behandelte Person kann davon betroffen sein. Daneben können weitere unerwünschte Reaktionen auftreten, die meist gut behandelbar sind. Betroffene sollten sich diesbezüglich an ihr Behandlungsteam wenden.16
Wie sieht die Begleittherapie aus?
Die bei Morbus Fabry infolge der Schädigungen an Geweben und Organen auftretenden Beschwerden können zusätzlich unabhängig von der eigentlichen Krankheitsursache behandelt werden. Beispielsweise können verschiedene Wirkstoffarten für die Linderung von chronischen oder phasenweise auftretenden Schmerzen eingesetzt werden.21,22 Spezialisierte Schmerztherapeut*innen sind Expert*innen auf diesem Gebiet und stellen eine wichtige Anlaufstelle für Betroffene dar.13
Funktionsstörungen der Nieren werden ebenfalls zusätzlich mit Medikamenten behandelt. Sind die Schäden jedoch so groß, dass die Nierenfunktion unzureichend ist, kann eine regelmäßige Dialyse oder eine Nierentransplantation erforderlich sein.21,22
Um Herzerkrankungen vorzubeugen bzw. zu behandeln, können Medikamente zur Blutdruckkontrolle sowie zur Reduktion von Fettstoffen (Lipiden) und zur Hemmung der Blutgerinnung nötig sein. Letztere dienen auch der Vorbeugung von Schlaganfällen.21,22
Auch für Beschwerden des Verdauungstraktes und der Atemwege gibt es medikamentöse Behandlungsmöglichkeiten. Bei einer Einschränkung des Hörvermögens können wiederum Hörgeräte helfen.22
Eine wichtige Rolle spielt zudem die psychologische Beratung und Behandlung, da bei Menschen mit Morbus Fabry nicht selten auch depressive Verstimmungen auftreten können.22 Hilfreiche Informationen hierzu sind unter psychosoziale Unterstützung zusammengefasst.
Welche Nebenwirkungen können bei der Begleittherapie auftreten?
Für die Behandlung der Symptome und Folgeerkrankungen kann eine Vielzahl von Wirkstoffen eingesetzt werden, die sich in ihrer Wirkweise stark unterscheiden. Jedes dieser Medikamente weist ein eigenes Spektrum an möglichen Nebenwirkungen auf. Hierzu berät das Behandlungsteam und der*die Apotheker*in.
Grundsätzlich gilt, dass bei der Erstellung des Therapieplans die Vorteile und möglichen Risiken einer Behandlung immer sorgfältig gegeneinander abgewogen werden sollten.
Wie kann die Lebensweise die Therapie unterstützen?
Patient*innen mit Morbus Fabry können mithilfe einiger einfacher Maßnahmen im Alltag den Erfolg der Behandlung positiv beeinflussen und ihr Wohlbefinden verbessern:13,22
Schmerz-Episoden kann durch Vermeidung von körperlicher Überanstrengung, Stress, Übermüdung und extremen Temperaturen vorgebeugt werden. Bei großer Hitze können Klimaanlagen, Kühlwesten und kühlende Gesichtssprays hilfreich sein. Zudem ist es wichtig, ausreichend zu trinken.
Beschwerden des Verdauungstrakts können durch kleinere, häufigere Mahlzeiten und einen erhöhten Verzehr von Ballaststoffen reduziert werden.
Die Nierenfunktion kann durch eine salz- und eiweißarme Ernährung unterstützt werden.
Zur Vorbeugung von Lungen- und Herzerkrankungen ist es wichtig, nicht zu rauchen.
Können Betroffene an Studien teilnehmen?
Um die Erkrankung Morbus Fabry besser zu verstehen und die Behandlungsmöglichkeiten zu optimieren, wird intensiv Forschung betrieben. Dazu gehört auch die Durchführung von klinischen Studien. Unter bestimmten Voraussetzungen ist es möglich, an einer solchen Studie teilzunehmen. Informationen hierzu bieten die spezialisierten Fabry-Zentren an.
- Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab 2018;124(3):189-203
- Paim-Marques L, de Oliveira RJ, Appenzeller S. Multidisciplinary management of fabry disease: current perspectives. J Multidiscip Healthc 2022;15:485-95
- Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30
- Beck M, Hughes D, Kampmann C, et al. Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: a Fabry Outcome Survey analysis. Mol Genet Metab Rep 2015;3:21-7
- Germain DP, Charrow J, Desnick RJ, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet 2015;52(5):353-8
- Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 2007;146(2):77-86
- Wilcox WR, Banikazemi M, Guffon N, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet 2004;75(1):65-74
- Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease. N Engl J Med 2001;345(1):9-16
- Germain DP, Waldek S, Banikazemi M, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol 2007;18(5):1547-57
- Wallace E, Goker-Alpan O, Alon S, et al. Safety and efficacy of pegunigalsidase alfa vs agalsidase beta on renal function in Fabry disease: 24-month results from the phase III randomized, double-blind BALANCE study. 7th Update on Fabry Disease, Würzburg,
- Borgwardt L, Feldt-Rasmussen U, Rasmussen AK, et al. Fabry disease in children: agalsidase-beta enzyme replacement therapy. Clin Genet 2013;83(5):432-8
- Schiffmann R, Martin RA, Reimschisel T, et al. Four-year prospective clinical trial of agalsidase alfa in children with Fabry disease. J Pediatr 2010;156(5):832-7, 7.e1
- Morbus Fabry Selbsthilfegruppe e.V. Morbus Fabry Broschüre. https://fabry-shg.org/informationsmaterial-eigenes/, abgerufen am: 28.09.2022
- Holida M, Bernat J, Longo N, et al. Safety and efficacy of pegunigalsidase alfa administered every 4 weeks in patients with Fabry disease: results from the phase 3, open-label, BRIGHT study. WORLD Symposium, San Diego, USA, 7.-11.2.2022
- Beck M, Gaedeke J, Martus P, et al. Infusionsbehandlung in häuslicher Umgebung--ein praktikabler Ansatz für chronisch Kranke? Neue Wege der Versorgung am Beispiel des Morbus Fabry. Dtsch Med Wochenschr 2013;138(46):2345-50
- Fachinfo-Service. 2022. www.fachinfo.de, abgerufen am: 28.10.2022
- Schiffmann R, Goker-Alpan O, Holida M, et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: a 1-year Phase 1/2 clinical trial. J Inherit Metab
- Lenders M, Brand E. Mechanisms of neutralizing anti-drug antibody formation and clinical relevance on therapeutic efficacy of enzyme replacement therapies in Fabry disease. Drugs 2021;81(17):1969-81
- Parenti G, Andria G, Valenzano KJ. Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol Ther 2015;23(7):1138-48
- Rote Liste. 2022. www.rote-liste.de, abgerufen am: 24.10.2022
- Mehta A, Hughes DA. Fabry disease. In: Adam MP, Everman DB, Mirzaa GM, et al. (Hrsg.), GeneReviews® [Internet]. University of Washington, Seattle. Copyright © 1993-2022, Seattle (WA), 2002 [Updated 2022]
- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 2018;123(4):416-27